Emma is the first author of an article published in Nature Communications about the existing differences among neurons expressing dopamine D2 receptors in the striatum. It is an international collaboration in which Albert and Eli also participated.

The striatum is a brain region involved in motor control, habit formation, decision-making, motivation and reinforcement, among other aspects, and its disfunction has been associated with many neurological and psychiatric disorders. One of the most important neurotransmitters in the striatum is dopamine, which exerts different functions depending on the kind of receptor it binds to.
This study, which is an international collaboration, focused on D2 receptors, and showed that, contrary to paradigm, not all D2 neurons within the striatum have the same molecular identify or function, but that their neuro-anatomical localization is key.
Using cutting-edge technologies, they analyzed mouse models to see what genes are expressed in D2 neurons from the two main areas of the striatum: the ventral striatum, consisting mainly of the nucleus accumbens, and the dorsal striatum, and revealed overwhelming differences among them. Thus, depending on their precise anatomical location, they express different kinds of proteins, changing neurons’ features and functions.
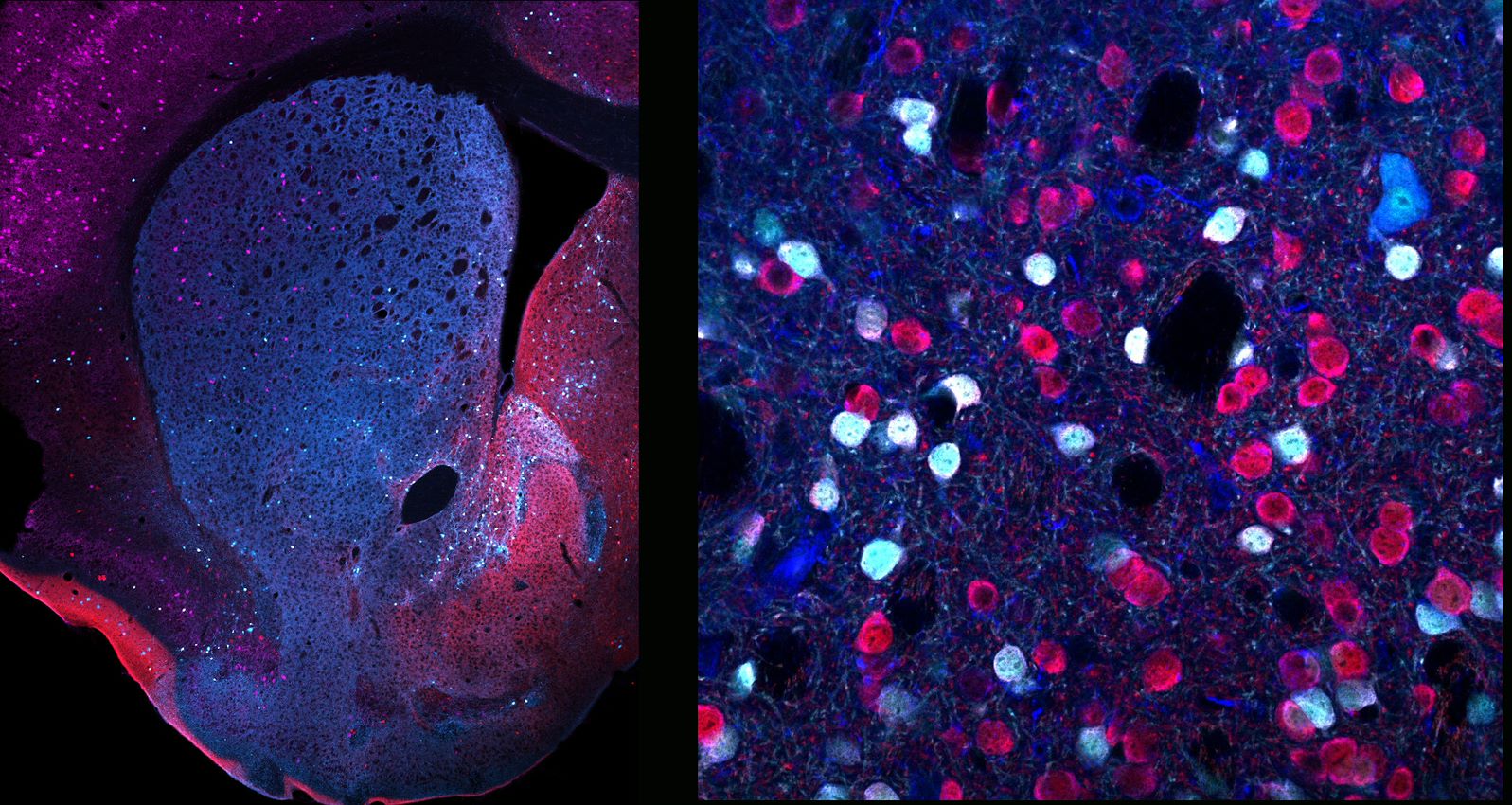
They also focused on a group of neurons mainly located in the accumbens, which express the protein WFS1, and studied the effects of deleting their D2 receptors. What they observed was a significant reduction in digging, an innate behavior used in many species to seek or hoard food, as shelter, or to hide away from predators, whose underlying neuronal mechanisms were still unknown. Additionally, the authors found that these animals present an exacerbated hyperlocomotor response when their dopamine levels are increased through amphetamine administration, suggesting a key role of D2 receptors from WFS1 neurons in the response to psychostimulants.
Overall, this study demonstrates that there is a huge complexity and functional specificity among D2 neuron subpopulations, and reveals the possibility to manipulate them specifically to better understand their functions, in both physiological and pathological contexts.